


![]()
Болезнь Гоше
Болезнь Гоше — редкое наследственное заболевание обмена веществ, которое относится к сфинголипидозам — одному из типов лизосомных болезней накопления.
Что такое болезнь Гоше?
Болезнь Гоше (названная в честь Филиппа Гоше, описавшего это заболевание в 1882 г.1) является сфинголипидозом, одним из типов лизосомных болезней накопления2. Сфинголипидозы представляют собой группу наследственных заболеваний, обусловленных генетическими нарушениями системы катаболизма сфинголипидов в лизосомах, которые приводят к накоплению нерасщепленных макромолекул в одном или нескольких органах3,4. Сфинголипиды, наряду с глицерофосфолипидами и холестерином, являются строительным материалом мембран эукариотических клеток3. Гликосфинголипиды образуются путем присоединения углеводной части к церамиду (N-ацилсфингозин)5. Гликосфинголипиды необходимы для жизнедеятельности многоклеточных организмов3,6,7.
Болезнь Гоше является наиболее распространенным типом сфинголипидозов и представляет собой хроническое системное наследственное заболевание, где органами-мишенями являются печень, селезенка, костный мозг и лимфатические узлы. В зависимости от возраста манифестации заболевания, клинических признаков, а также наличия и скорости прогрессирования неврологических расстройств выделяют различные типы болезни Гоше8:
В чем причина болезни Гоше?
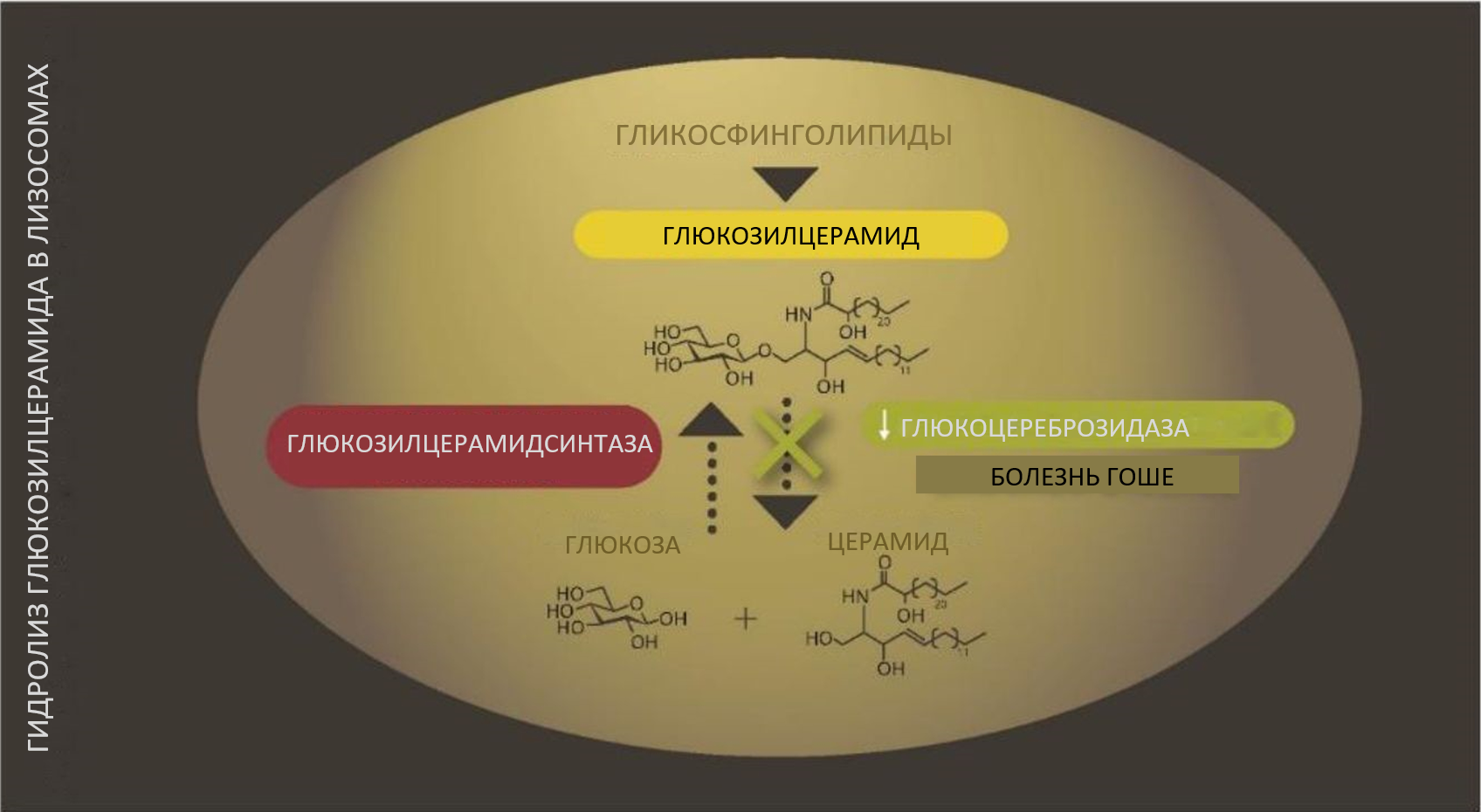
Причиной болезни Гоше являются мутации гена GBA1, расположенного в 1-й хромосоме (см. раздел «Генетические аспекты болезни Гоше»). Мутации этого гена приводят к значительному снижению активности лизосомального фермента глюкоцереброзидазы, которая гидролизует глюкозилцерамид с образованием церамида и глюкозы2 (рис. 1). Глюкозилцерамид — это широко распространенный гликосфинголипид, центральная структура глобозидных и ганглиозидных комплексов, которые являются важными компонентами клеточных мембран, встроенных рецепторов и липидных рафтов9,10.
Рис. 1.
Гидролиз глюкозилцерамида глюкоцереброзидазой в лизосоме2
При болезни Гоше дефицит глюкоцереброзидазы приводит к накоплению глюкозилцерамида в лизосомах (рис. 2). Затем глюкозилцерамиды образуют фибриллярные агрегаты, накапливающиеся в макрофагах, в результате чего цитоплазма клеток приобретает характерный вид «скомканной папиросной бумаги». Эти клетки называются клетками Гоше, которые способны инфильтрировать многие органы, включая костный мозг, селезенку и печень, что обусловливает клинические проявления болезни Гоше2. Например, инфильтрация костного мозга клетками Гоше уменьшает количество стволовых клеток и может вызвать фиброз, инфаркт, некроз и рубцевание других тканей. Однако наличие клеток Гоше не объясняет полностью патофизиологические механизмы болезни Гоше. Несмотря на крупный размер, клетки Гоше не являются инертными, они метаболически активны и могут синтезировать и секретировать белковые молекулы, регулирующие другие патофизиологические процессы11. У пациентов с болезнью Гоше были выявлены показатели активации макрофагов (например, лиганд хемокина CCL8 [CCL18], кластер дифференцировки 163 (CD 163), хитотриозидаза, гранулоцитарно-макрофагальный колониестимулирующий фактор (ГМ-КСФ) и растворимая форма CD1412–14. Кроме того, были получены научные данные, позволяющие предположить, что провоспалительные макрофаги окружают клетки Гоше и мигрируют в органы-мишени11. Вместе с тем у пациентов с болезнью Гоше повышается уровень глюкозилсфингозина (лизо-Gb1), метаболита глюкозилцерамида, который рассматривают как биомаркер заболевания15,16. При длительной подкожной инфузии глюкозилсфингозина (лизо-Gb1) мышам, установлено, что его концентрация повышается до значений, которые наблюдаются у пациентов со среднетяжелой и тяжелой формами болезни Гоше17,18. Более того, после инфузии гликозилсфингозина (лизо-Gb1) интактным мышам отмечено, что у них развиваются фенотипические изменения крови и селезенки, аналогичные изменениям, наблюдаемым у мышей с моделью болезни Гоше 1 типа, что указывает на роль глюкозилсфингозина (лизо-Gb1) в патофизиологии болезни Гоше17.
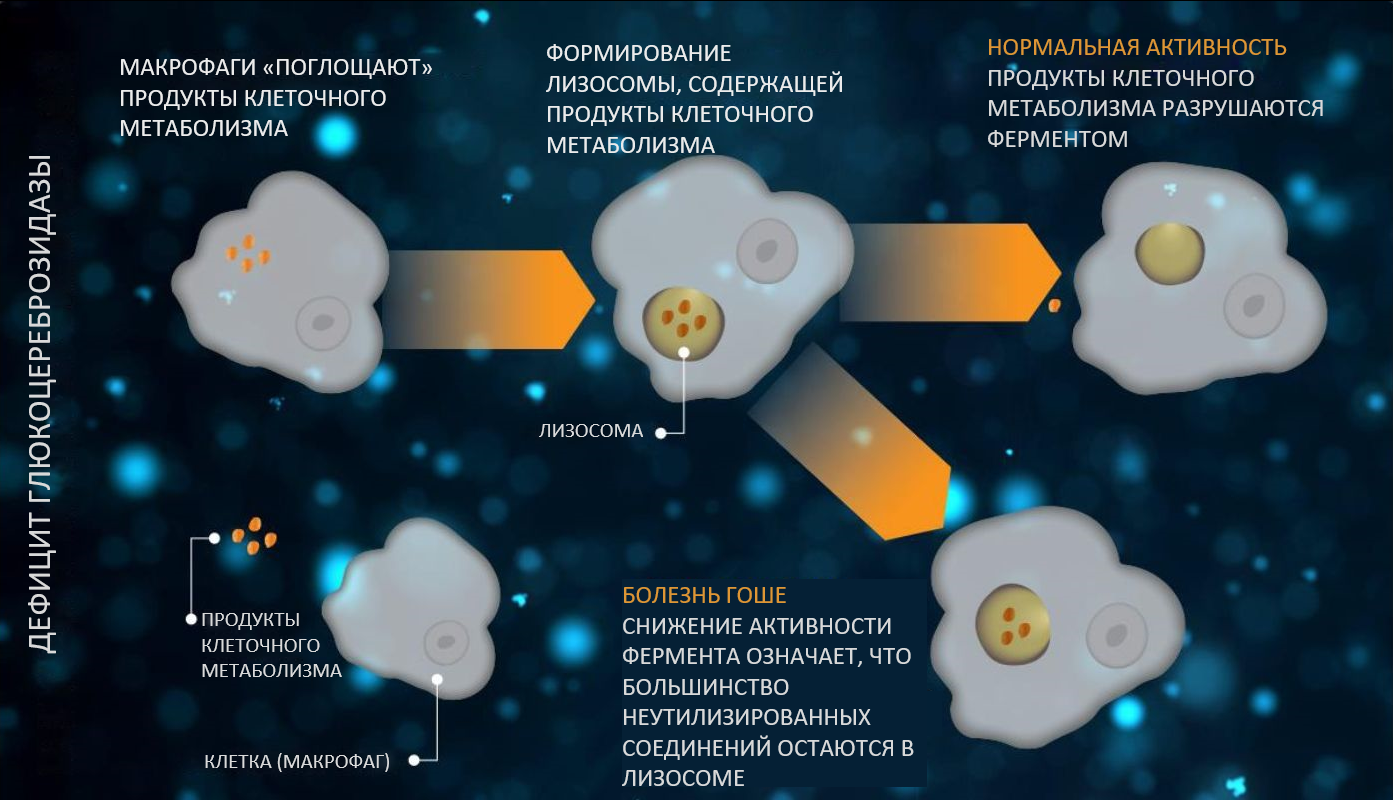
Рис. 2.
При болезни Гоше снижение активности глюкоцереброзидазы приводит к накоплению глюкозилцерамида в лизосомах и увеличению макрофагов19
- Gaucher PCE. De l'épithélioma primitif de la rate. Hypertrophie idiopathique de la rate sans leucémie. 1882. Available at: https://archive.org/details/BIUSante_TPAR1882x031/mode/2up. Accessed September 2020.
Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 2017; 18: 441.
Kolter T, Sandhoff K. Sphingolipid metabolism diseases. Biochim Biophys Acta 2006; 1758: 2057-2079.
- Raas-Rothschild A, Pankova-Kholmyansky I, Kacher Y, et al. Glycosphingolipidoses: beyond the enzymatic defect. Glycoconj J 2004; 21: 295-304.
- Watts RW. A historical perspective of the glycosphingolipids and sphingolipidoses. Philos Trans R Soc Lond B Biol Sci 2003; 358: 975-983.
- Yamashita T, Wada R, Sasaki T, et al. A vital role for glycosphingolipid synthesis during development and differentiation. Proc Natl Acad Sci U S A 1999; 96: 9142-9147.
- Yang LJ, Zeller CB, Shaper NL, et al. Gangliosides are neuronal ligands for myelin-associated glycoprotein. Proc Natl Acad Sci U S A 1996; 93: 814-818.
- Baris HN, Cohen IJ, Mistry PK. Gaucher disease: the metabolic defect, pathophysiology, phenotypes and natural history. Pediatr Endocrinol Rev 2014; 12 Suppl 1: 72-81.
- Rosenbloom BE, Weinreb NJ. Gaucher disease: a comprehensive review. Crit Rev Oncog 2013; 18: 163-175.
- Messner MC, Cabot MC. Glucosylceramide in humans. Adv Exp Med Biol 2010; 688: 156-164.
- Bussink AP, van Eijk M, Renkema GH, et al. The biology of the Gaucher cell: the cradle of human chitinases. Int Rev Cytol 2006; 252: 71-128
- Brinkman J, Wijburg FA, Hollak CE, et al. Plasma chitotriosidase and CCL18: early biochemical surrogate markers in type B Niemann-Pick disease. J Inherit Metab Dis 2005; 28: 13-20.
- Boven LA, van Meurs M, Boot RG, et al. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am J Clin Pathol 2004; 122: 359-369.
- Hollak CE, Evers L, Aerts JM, et al. Elevated levels of M-CSF, sCD14 and IL8 in type 1 Gaucher disease. Blood Cells Mol Dis 1997; 23: 201-212.
- Orvisky E, Park JK, LaMarca ME, et al. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab 2002; 76: 262-270.
- Dekker N, van Dussen L, Hollak CE, et al. Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood 2011; 118: e118-e127.
- Lukas J, Cozma C, Yang F, et al. Glucosylsphingosine causes hematological and visceral changes in mice–evidence for a pathophysiological role in Gaucher disease. Int J Mol Sci 2017; 18: 2192.
- Rolfs A, Giese AK, Grittner U, et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS One 2013; 8: e79732.
- Mehta A. Epidemiology and natural history of Gaucher's disease. Eur J Intern Med 2006; 17 Suppl: S2-S5.
Признаки и симптомы болезни Гоше
Каковы признаки и симптомы болезни Гоше?
Частичное совпадение типов болезни Гоше может затруднить диагностику. Однако определение клинических фенотипов при каждом типе болезни Гоше важно из-за значительных различий в клинических исходах и прогнозах заболевания
Генетические аспекты болезни Гоше
Как наследуется болезнь Гоше?
Болезнь Гоше — аутосомно-рецессивное заболевание, вызванное мутацией гена глюкоцереброзидазы GBA1.
Тактика ведения пациентов
Лечение болезни Гоше требует пациент-ориентированного подхода. Разработаны препараты для ферментозаместительной и субстрат-редуцирующей терапии болезни Гоше.
Болезнь Гоше 1 типа -
Что такое болезнь Гоше 1-го типа ?
Болезнь Гоше 1-го типа является наиболее распространенной формой заболевания и может манифестировать в любом возрасте. Клиническими проявлениями болезни Гоше 1-го типа являются гепатомегалия, спленомегалия, тромбоцитопения, анемия и поражение костей.
Болезнь Гоше 2 типа
Что такое болезнь Гоше 2 типа?
Болезнь Гоше 2 типа обычно развивается у детей в возрасте до 6 месяцев и сопровождается тяжелым поражением нервной системы. Помимо проявлений, связанных с поражением нервной системы, основными признаками болезнь Гоше 2 типа - являются спленомегалия, тромбоцитопения и поражение легких.
Болезнь Гоше 3 типа -
Что такое болезнь Гоше 3 типа?
Болезнь Гоше 3-го типа развивается в детстве и медленно прогрессирует в течение всей жизни. Кроме висцеральных, гематологических и скелетных проявлений, у пациентов с болезнью Гоше 3-го типа наблюдается поражение нервной системы. У пациентов с болезнью Гоше 3-го типа степень поражения нервной системы может варьировать. Первым проявлением может быть межъядерная офтальмоплегия.